|
|
 发表于 2022-12-1 13:08:23
|
显示全部楼层
发表于 2022-12-1 13:08:23
|
显示全部楼层
这次我们来探索各类肿瘤与对照的全局(如mRNA,lncRNA,miRNA,甲基化,蛋白)水平的差异情况(差异分析流程),也就是各类型肿瘤的turmorl和normal的差异分析,用的包是UCSCXenaTools,UCSCXenaTools包以往的介绍见:通过R包UCSCXenaTools链接UCSC的XENA浏览器来探索TCGA等公共数据、UCSCXenaTools介绍。
设置
rm(list=ls())options(stringsAsFactors = F)library(data.table)library(dplyr)library(stringi)#install.packages("UCSCXenaTools")library(UCSCXenaTools)通过标准分析流程五步走
生成 XenaHub 对象
#UCSCXenaTools 使用包内置数据集 XenaData 辅助生成 XenaHub 对象,这个数据集记录了当前所有数据集的信息。data(XenaData) head(XenaData)#> # A tibble: 6 x 17#> XenaHosts XenaHostNames XenaCohorts XenaDatasets SampleCount DataSubtype#> <chr> <chr> <chr> <chr> <int> <chr> #> 1 https://… publicHub Breast Can… ucsfNeve_pu… 51 gene expre…#> 2 https://… publicHub Breast Can… ucsfNeve_pu… 57 phenotype #> 3 https://… publicHub Glioma (Ko… kotliarov20… 194 copy number#> 4 https://… publicHub Glioma (Ko… kotliarov20… 194 phenotype #> 5 https://… publicHub Lung Cance… weir2007_pu… 383 copy number#> 6 https://… publicHub Lung Cance… weir2007_pu… 383 phenotype #> # … with 11 more variables: Label <chr>, Type <chr>,#> # AnatomicalOrigin <chr>, SampleType <chr>, Tags <chr>, ProbeMap <chr>,#> # LongTitle <chr>, Citation <chr>, Version <chr>, Unit <chr>,#> # Platform <chr>#从&#39;XenaData&#39;生成XenaHub对象,取子集xe = XenaGenerate(subset = XenaHostNames == &#34;gdcHub&#34;)过滤数据
xe2 = XenaFilter(xe, filterDatasets = &#34;htseq_counts&#34;) xe2@datasets# [1] &#34;TCGA-BLCA.htseq_counts.tsv&#34; &#34;TCGA-LUSC.htseq_counts.tsv&#34; # [3] &#34;TCGA-ESCA.htseq_counts.tsv&#34; &#34;TARGET-RT.htseq_counts.tsv&#34; # [5] &#34;MMRF-COMMPASS.htseq_counts.tsv&#34; &#34;TCGA-MESO.htseq_counts.tsv&#34; # [7] &#34;TCGA-LUAD.htseq_counts.tsv&#34; &#34;TCGA-STAD.htseq_counts.tsv&#34; # [9] &#34;TCGA-TGCT.htseq_counts.tsv&#34; &#34;TCGA-UVM.htseq_counts.tsv&#34; # [11] &#34;TCGA-PAAD.htseq_counts.tsv&#34; &#34;TCGA-GBM.htseq_counts.tsv&#34; # [13] &#34;TCGA-COAD.htseq_counts.tsv&#34; &#34;TARGET-NBL.htseq_counts.tsv&#34; # [15] &#34;TCGA-OV.htseq_counts.tsv&#34; &#34;TCGA-THYM.htseq_counts.tsv&#34; # [17] &#34;TCGA-BRCA.htseq_counts.tsv&#34; &#34;TCGA-UCEC.htseq_counts.tsv&#34; # [19] &#34;TCGA-UCS.htseq_counts.tsv&#34; &#34;TARGET-WT.htseq_counts.tsv&#34; # [21] &#34;TARGET-AML.htseq_counts.tsv&#34; &#34;TCGA-CHOL.htseq_counts.tsv&#34; # [23] &#34;TARGET-OS.htseq_counts.tsv&#34; &#34;TCGA-KIRC.htseq_counts.tsv&#34; # [25] &#34;TCGA-ACC.htseq_counts.tsv&#34; &#34;TCGA-LGG.htseq_counts.tsv&#34; # [27] &#34;TCGA-KIRP.htseq_counts.tsv&#34; &#34;TCGA-KICH.htseq_counts.tsv&#34; # [29] &#34;TCGA-THCA.htseq_counts.tsv&#34; &#34;TARGET-ALL-P3.htseq_counts.tsv&#34;# [31] &#34;TCGA-LIHC.htseq_counts.tsv&#34; &#34;TCGA-HNSC.htseq_counts.tsv&#34; # [33] &#34;TCGA-CESC.htseq_counts.tsv&#34; &#34;TCGA-READ.htseq_counts.tsv&#34; # [35] &#34;TCGA-PCPG.htseq_counts.tsv&#34; &#34;TARGET-CCSK.htseq_counts.tsv&#34; # [37] &#34;TCGA-SARC.htseq_counts.tsv&#34; &#34;TCGA-DLBC.htseq_counts.tsv&#34; # [39] &#34;TCGA-PRAD.htseq_counts.tsv&#34; &#34;TCGA-LAML.htseq_counts.tsv&#34; # [41] &#34;TCGA-SKCM.htseq_counts.tsv&#34;检索数据
xe2_query = XenaQuery(xe2)head(xe2_query)# hosts datasets# 1 https://gdc.xenahubs.net TCGA-BLCA.htseq_counts.tsv# 2 https://gdc.xenahubs.net TCGA-LUSC.htseq_counts.tsv# 3 https://gdc.xenahubs.net TCGA-ESCA.htseq_counts.tsv# 6 https://gdc.xenahubs.net TCGA-MESO.htseq_counts.tsv# 7 https://gdc.xenahubs.net TCGA-LUAD.htseq_counts.tsv# 8 https://gdc.xenahubs.net TCGA-STAD.htseq_counts.tsv# url# 1 https://gdc.xenahubs.net/download/TCGA-BLCA.htseq_counts.tsv.gz# 2 https://gdc.xenahubs.net/download/TCGA-LUSC.htseq_counts.tsv.gz# 3 https://gdc.xenahubs.net/download/TCGA-ESCA.htseq_counts.tsv.gz# 6 https://gdc.xenahubs.net/download/TCGA-MESO.htseq_counts.tsv.gz# 7 https://gdc.xenahubs.net/download/TCGA-LUAD.htseq_counts.tsv.gz# 8 https://gdc.xenahubs.net/download/TCGA-STAD.htseq_counts.tsv.gzsave(xe2_query, file = &#34;03-DEG/xe2_query.RData&#34;)下载和导入数据放在画图的循环里。
循环画图
load(&#34;03-DEG/xe2_query.RData&#34;)xe2_query = xe2_query[substr(xe2_query$datasets,1,4)== &#39;TCGA&#39;,] #去掉其它数据库来源的数据,因为分组规则不一样(不一定按01-11分组),后续画图会出错destdir = &#34;03-DEG/xena_TCGA_data&#34; #下载数据存入的文件夹library(limma)for (i in 1:length(xe2_query$datasets) ) { #i = 14 xe2_query2 = xe2_query[i,] #下载数据,已经下载了会读入 xe2_download = XenaDownload(xe2_query2, destdir = destdir, trans_slash = TRUE)#导入数据 expr = XenaPrepare(xe2_download) expr$Ensembl_ID=stri_sub(expr$Ensembl_ID,1,15)##保留前15位,去掉小数点 row.names(expr)=expr$Ensembl_ID expr2 =expr[,-1] row.names(expr2)=expr$Ensembl_ID expr2=normalizeBetweenArrays(expr2) #转换一下 id library(tinyarray) expr2 = trans_exp(expr2) #tumor和normal分组信息 group = ifelse(as.numeric(substr(colnames(expr2),14,15)) < 10,&#39;tumor&#39;,&#39;normal&#39;) #group = ifelse(as.numeric(substr(colnames(expr2),nchar(colnames(expr2))-2,nchar(colnames(expr2))-1)) < 10,&#39;tumor&#39;,&#39;normal&#39;) group <- factor(group, levels = c(&#39;normal&#39;,&#39;tumor&#39;)) #因为有一些癌症数据集是没有normal样本的,所以要做一个判断,且使用normal样本大于15的数据 if (sum(group==&#34;normal&#34;)>15) { #if (&#34;normal&#34; %in% group) { #转录组数据用limma做差异分析 library(limma) dge <- edgeR::DGEList(counts=expr2) dge <- edgeR::calcNormFactors(dge) design=model.matrix(~factor( group )) fit = voom(dge,design, normalize=&#34;quantile&#34;) fit=lmFit(expr2,design) fit=eBayes(fit) options(digits = 4) #设置全局的数字有效位数为4 deg = topTable(fit,coef=2,adjust=&#39;BH&#39;, n=Inf) #获取具体的癌症名字 cancer = xe2_query2$datasets cancer = gsub(&#34;TCGA-&#34;,&#34;&#34;,cancer) cancer = gsub(&#34;.htseq_counts.tsv&#34;,&#34;&#34;,cancer) cancer save(deg, file=paste0(&#34;03-DEG/&#34;,cancer,&#34;_limma_deg.Rdata&#34;)) #取上下调显著的前10个基因画图 logFC_t=1 P.Value_t = 0.05 k1 = (deg$P.Value < P.Value_t)&(deg$logFC < -logFC_t) k2 = (deg$P.Value < P.Value_t)&(deg$logFC > logFC_t) deg <- mutate(deg,change = ifelse(k1,&#34;down&#34;,ifelse(k2,&#34;up&#34;,&#34;stable&#34;))) library(dplyr) dat2 = deg %>% filter(change!=&#34;stable&#34;) %>% arrange(logFC) #把上下调基因按logFC排序 cg = c(head(rownames(dat2),5), tail(rownames(dat2),5)) #火山图 library(EnhancedVolcano) EnhancedVolcano(deg, lab = rownames(deg), x = &#39;logFC&#39;, y = &#39;P.Value&#39;, pCutoff = 0.05, FCcutoff = 1, labSize = 5, selectLab = cg, drawConnectors = TRUE #EnhancedVolcano显示基因名会默认重叠就不显示,加上这个参数才会错开显示 ) ggsave(width = 10, height = 8, filename = paste0(&#34;03-DEG/&#34;,cancer,&#34;_limma_deg.png&#34;)) }}有14个癌症的normal数据大于15个画出了图,放三个看看:
COAD
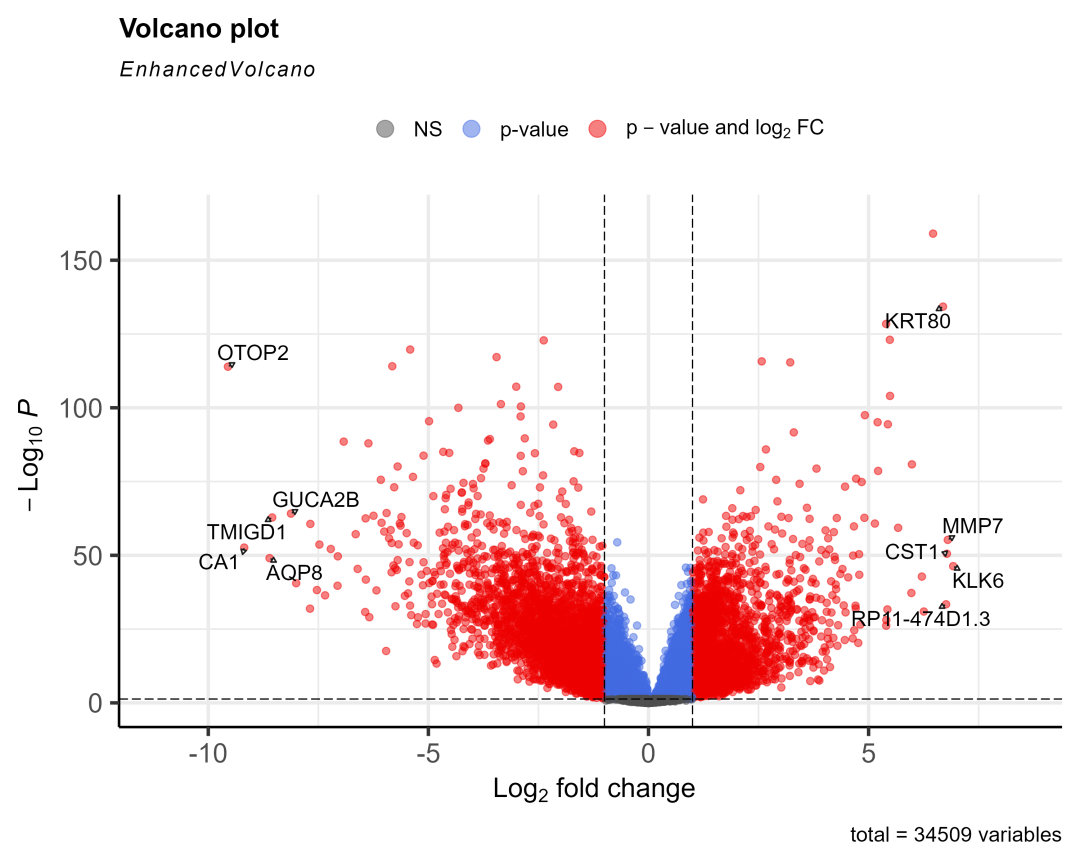
STAD
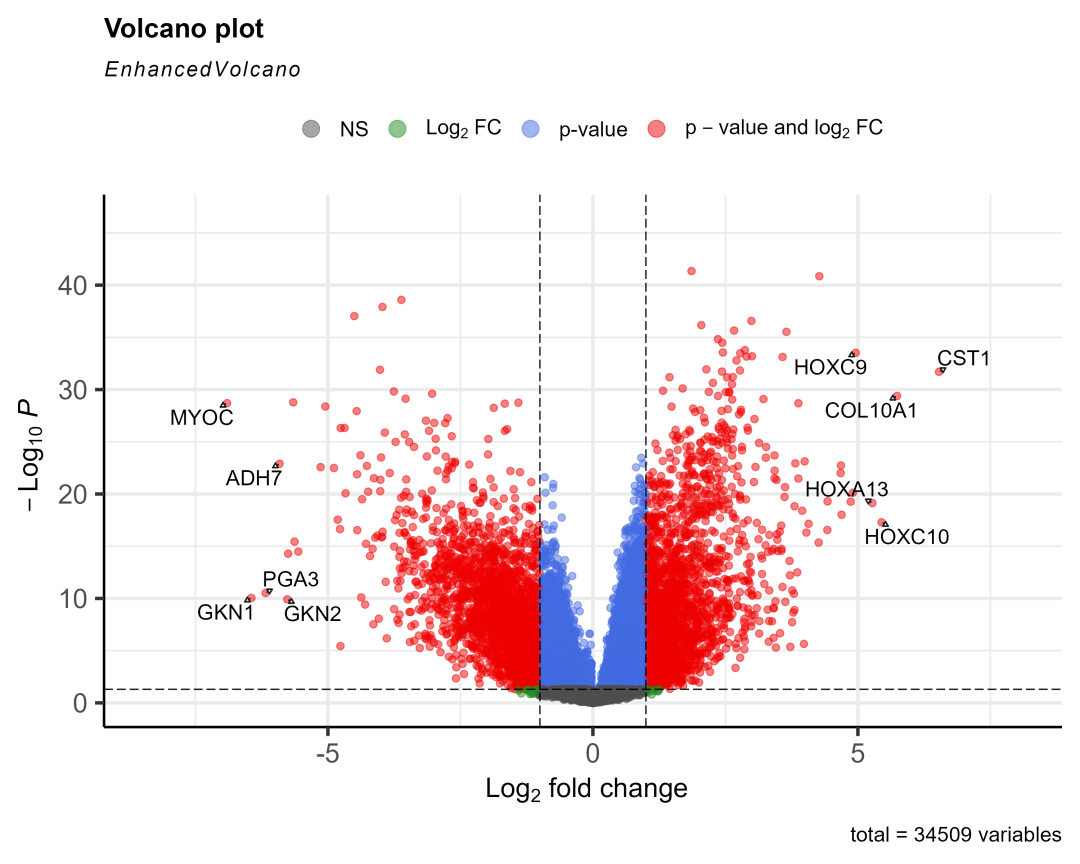
THCA
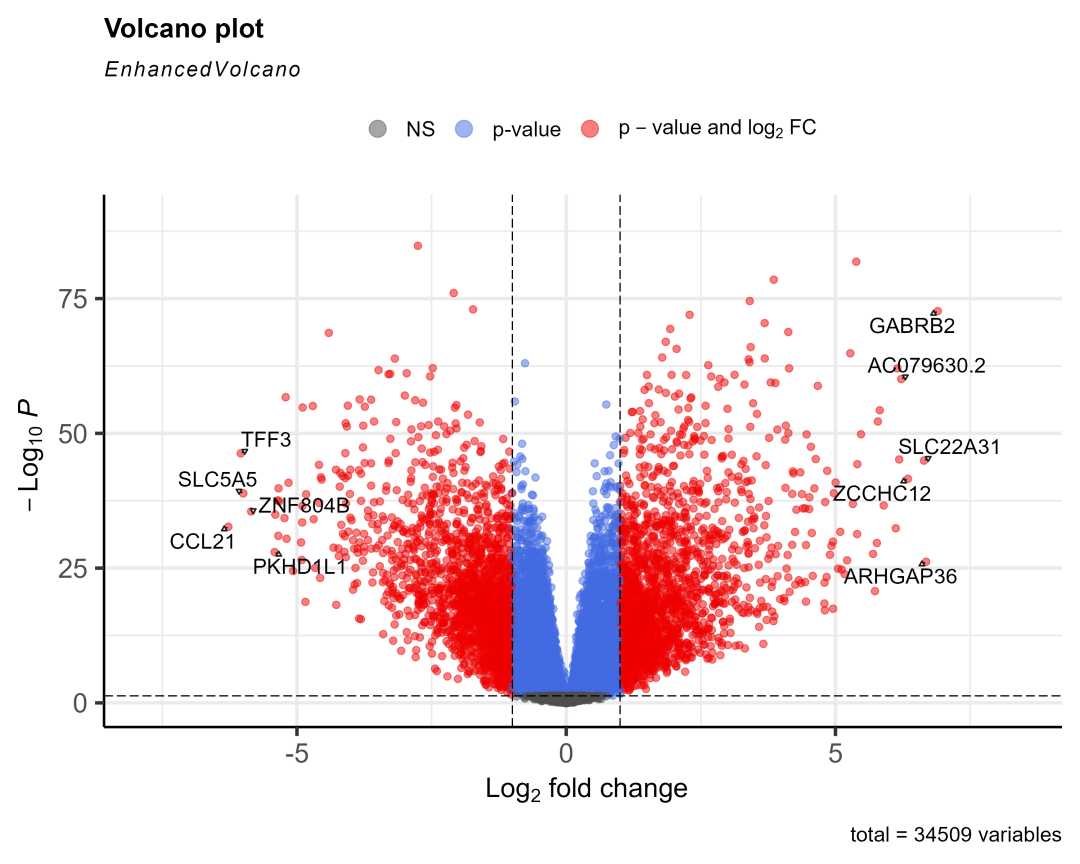 |
|

